I recently had the opportunity to collaborate with Kevin Hall, PhD and Rudy Leibel, MD on a commentary in JAMA Internal Medicine (1). It was fun for me to work with two researchers who I respect tremendously. Hall’s energy balance modeling work has brought important new insights to the obesity research field and Leibel is, well, the co-discoverer of leptin. And he has done as much as anyone else to help us understand how this hormone works in humans.
Our commentary is titled “The Carbohydrate-Insulin Model of Obesity Is Difficult to Reconcile With Current Evidence”, and we wrote it in response to a review paper in the same issue written by David Ludwig, MD, PhD and Cara Ebbeling, PhD, titled “The Carbohydrate-Insulin Model of Obesity: Beyond ‘Calories In, Calories Out'” (2). In this paper, Ludwig and Ebbeling lay out their argument for the carbohydrate-insulin model (CIM), as they refer to it. This is nearly identical to the model Gary Taubes advocates: obesity is primarily caused by the ability of carbohydrate to increase insulin secretion, which reduces levels of circulating fuels (glucose and free fatty acids), shunts fat into fat cells, and makes us fat, hungry, and sluggish. According to this model, high calorie intake and low calorie expenditure are a result of expanding fat tissue, not its cause. Ludwig and Ebbeling focus particularly on glycemic load, which is the degree to which diet impacts blood glucose. In my view, this review paper is the strongest defense of the model currently available.
While Ludwig and Ebbeling had ample space to develop their arguments, we were limited to 1,200 words and 8 references. So I’m going to add to our commentary here, expanding on some points and adding others. This post reflects my views and not necessarily those of Hall and Leibel, who were not involved in writing it.
I don’t want to be on the wrong side of history, and one way to do that is to make overly confident and categorical predictions. I still think it’s plausible that some insulin-related variable could be involved in obesity and/or fat loss, particularly 1) insulin resistance in energy-regulating circuits in the brain, and/or 2) blood glucose levels between meals, or some other signal of glucose availability. What I think is very unlikely to be correct is the hypothesis articulated by Ludwig, Ebbeling, and Taubes: the primary cause of obesity is carbohydrate-stimulated insulin acting on fat cells.
That said, I want to be clear that I think certain forms of carbohydrate are part of the explanation for obesity, and low-carbohydrate diets do cause fat loss in most people, with greater carbohydrate restriction typically resulting in greater fat loss. Like most diets, low-carbohydrate diets aren’t very effective against obesity in the average person, but they do have some effectiveness and they are certainly a valid tool in the toolbox. They may also be particularly useful for managing diabetes, although long-term outcomes remain uncertain.
Mischaracterization of opposing viewpoints
Ebbeling and Ludwig begin their JAMA piece by citing a recent Endocrine Society scientific statement written by leading obesity researchers, including Rudy Leibel and my mentor Mike Schwartz, MD (3). Ebbeling and Ludwig present it as an example of something they call the “Conventional Model”, which holds that calorie intake, expenditure, and body fatness are not biologically regulated and all we have to do is fire up our willpower and “eat less, move more” to conquer obesity. This is the opposite of what the Endocrine Society paper actually says, and our first clue is the fact that it’s published on behalf of the Endocrine Society in the journal Endocrine Reviews– both organizations dedicated to the study of hormones.
Schwartz, and most of the other authors, have built their careers studying the mechanisms that regulate calorie intake, expenditure, and body fatness. Schwartz has been battling the “Conventional Model” since the 1980s, as I detail in my book The Hungry Brain. Here’s an illustrative quote from my interview with him (page 145):
I started my fellowship in 1987 and was immediately indoctrinated into the idea that there is an adiposity control system. What I was working on was considered way out of the mainstream at the time. Everyone just assumed that obesity is a problem where people eat too much, and if they could control their eating and be normal, they wouldn’t have this problem.
Schwartz and others in his field proceeded to spend 30 years debunking this assumption. This field is the very reason the Conventional Model has lost steam over the last three decades. This is the field that discovered leptin and satiety hormones, showed that hormones and specific brain circuits regulate appetite and body fatness, and overturned the notion that body fatness is just about how much we happen to decide to eat and move (note that this is perfectly compatible with the prevailing view that when calorie intake is held constant, all foods are similarly fattening in humans).
Here’s another illustrative quote, this time from the abstract of the Endocrine Society scientific statement (3):
Growing evidence suggests that obesity is a disorder of the energy homeostasis system, rather than simply arising from the passive accumulation of excess weight.
It’s beyond my understanding how a person could read this paper and interpret it as a defense of the Conventional Model, or continue to believe that the Conventional Model is the primary alternative to the CIM.
In fact, our choice of models is not between the CIM and the Conventional Model, and I’m starting to feel like a broken record pointing this out. The Endocrine Society statement presents a third model, not mentioned by Ludwig and Ebbeling, that is neither the naive and oversimplified Conventional Model nor the CIM. It happens to be widely supported within the scientific community. It acknowledges many influences on body fatness, particularly the brain circuits that regulate food intake, calorie expenditure, and body fatness in response to environmental and internal signals (3). If Ludwig and Ebbeling want to convince the scientific community that their model is better than what currently exists, they should be arguing against the prevailing model, not a straw man that has been obsolete for quite some time.
Dietary fat can be fattening
This is a simple observation that is hard to reconcile with the CIM. Researchers have known for decades that adding fat to food tends to increase body fatness in animals. I have seen Olympics-worthy intellectual gymnastics to try to rationalize away this fact, but as I will show, the conclusion is impossible to escape. Here is a quote from a review paper on this topic (4): “With few exceptions, obesity is induced by high-fat diets in monkeys, dogs, pigs, hamsters, squirrels, rats, and mice.” This paper reviews many studies in these species suggesting that higher fat, usually at the expense of carbohydrate, increases body fatness.
In my own research, we used refined diets that were 60 percent fat and 20 percent carbohydrate to induce obesity in normal rats and mice– and they were very effective! In my hands, these diets caused detectable fat gain in three days, more than doubled body fat content in two weeks, and increased body fat by sixfold in 20 weeks (5, 6). The weight and fat gain on these diets is rapid, massive, and unmistakable. In fact, fat gain on refined high-fat diets occurs even when calorie intake isn’t allowed to increase, suggesting that they impact both calorie intake and expenditure (7). In their paper, Ludwig and Ebbeling suggest that only the CIM can explain the phenomenon of fat gain without increased calorie intake in rodents, but this is clearly not the case– my field has been aware of this in the context of high-fat diets and brain lesions for a long time.
These diets aren’t just high in fat of course. They’re based on refined ingredients and they usually contain carbohydrate (e.g., 20% of calories) including sugar (e.g., 7% of calories). Aha, you may say! The refined carbohydrate is why the diet is fattening, not the fat! Researchers have already tested that supposition by comparing this diet to a low-fat version in which the fat is mostly replaced by refined carbohydrate. Although the low-fat version can be fattening under some conditions (8), it isn’t as fattening as the high-fat version, and often it isn’t fattening at all. Here is a graph from a recently published study by Matthew Dalby, PhD, and colleagues, comparing fat mass gains over 8 weeks of free access to unrefined low-fat (black), refined low-fat (blue), and refined high-fat (red) food in mice (9):
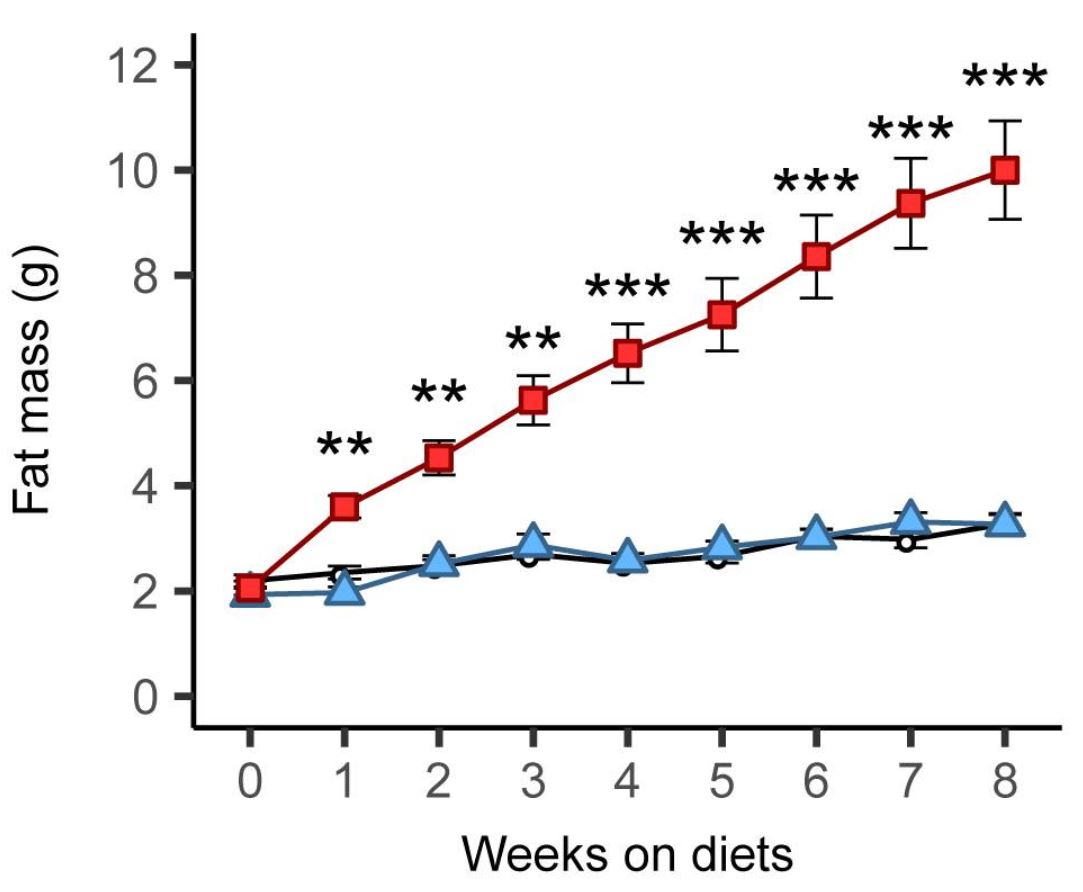
There was no difference between refined and unrefined low-fat diets, but the refined high-fat diet led to rapid and marked fat gain. This finding has been replicated independently, including in rats (10). [Update 7/18: a recent study using 29 diets and 5 strains of mice strongly supports the conclusion that dietary fat is much more fattening than carbohydrate, including sugar, in mice]
Refined carbohydrate and sugar are probably part of the reason why refined high-fat diets are fattening in animals, but there is no escaping the conclusion that the fat itself plays a key role. This simple observation is hard to reconcile with the CIM as articulated by Ludwig/Ebbeling/Taubes.
How about in humans? So far, two controlled overfeeding studies have compared diets rich in fat vs. carbohydrate and measured body fat gains (11, 12). Both studies reported that overfeeding with a high-fat diet produced the same, or even slightly greater, body fat gain than overfeeding with a high-carbohydrate diet of equal calories. Clearly, an excess calorie of fat gets into fat tissue as effectively as an excess calorie of carbohydrate, regardless of their effects on insulin.
How about when calorie intake isn’t controlled? Under these conditions, volunteers offered a high-fat diet tend to overeat and gain fat, just like in the animal studies discussed above (13, 14, 15, 16). Note that these studies didn’t use low-carbohydrate diets; they used high-fat diets that were unrestricted in carbohydrate, which is presumably why the results differ from low-carbohydrate diet studies in which people tend to eat fewer calories and lose fat.
Historically, many cultures ate high-carbohydrate (and high-glycemic-load) diets and were lean
This is another simple observation that is difficult to reconcile with the CIM. More precisely, it is a collection of many observations, because there have been, and continue to be, countless lean high-carbohydrate cultures. Ludwig has responded to this in the past, arguing that 1) the data are of insufficient quality to draw conclusions, 2) these people were lean because they were on the verge of starvation, and 3) they were protected by high levels of physical activity (17).
I’ll address these in turn, starting with the assertion that the data are of insufficient quality. The quality of data vary across observations, but some are quite high. For example, in 1990, Staffan Lindeberg, MD, PhD conducted a detailed survey of the diet, lifestyle, and health of the residents of Kitava, a Melanesian island scarcely touched by industrialization. He found a diet based on starchy plant foods (African yam, sweet potato, taro, cassava), fruit, vegetables, seafood, and coconut, with 69 percent of calories coming from carbohydrate (compared to ~49% in the US currently) (18).
Lindeberg described the Kitavans as “characterized by extreme leanness (despite food abundance)” (18). And the data back it up. Of the 247 Kitavans Lindeberg examined, none had obesity, and only a few were on the cusp of overweight (18). They did not gain weight with advancing age. There was no diabetes. Lindeberg described to me how the Kitavans had so much food they would let much of it rot or feed it to their animals. This is not to say that they never experienced food shortages– like all traditionally-living cultures, they probably did sometimes. However, they were not experiencing food shortage at the time of Lindeberg’s visit, and this is obvious in the photos he took of them (photos courtesy of Staffan Lindeberg):



Lindeberg actually did observe two Kitavan men who had borderline obesity. He describes them in his book Food and Western Disease (p. 119):
At the time of our survey in Kitava, we observed two cases of abdominal obesity, both in urbanised male migrants who had grown up in Kitavan and now came for a visit. We managed to examine one of them, a businessman aged 44 who differed in four variables from all other adults regardless of sex: he had the highest [body mass index], the highest waist-to-hip ratio, the highest diastolic blood pressure, and the highest PAI-1 in blood plasma… The most obvious difference in his lifestyle, as compared with non-migrant Kitavans, was the adoption of Western dietary habits.
This suggests that Kitavans were not genetically protected from fat gain and its metabolic consequences– they were protected by their diet/lifestyle, despite its high glycemic load. When they adopt an industrialized diet, which happens to be higher in fat and lower in carbohydrate, they become fat just like the rest of us.
Were the Kitavans protected from obesity by very high levels of physical activity? Lindeberg tested this hypothesis, and here’s what he found (18):
The amount of physical activity is estimated at 1.7 multiples of the basal metabolic rate, which is slightly higher than in sedentary Western populations.
In Food and Western Disease, he states that this corresponds to Westerners with “a moderate amount of physical activity on the job and during leisure time” (p. 84). I have little doubt that regular physical activity is part of the reason why Kitavans were lean and healthy, but their physical activity level was nothing extreme.
The book Western Diseases: Their Emergence and Prevention is primarily a collection of field studies from around the world that document the transition between traditional and modern lifestyles, and its health impacts. In most (not all) cases, the transition was from a very-low-fat, very-high-starch, high-glycemic-load diet to a more fatty diet, and this was invariably accompanied by increased rates of obesity and chronic disease. Certainly, the shift from carbohydrate to fat wasn’t the only change and we can’t attribute increasing body fatness to it exclusively. Concurrently, many of these cultures experienced increased consumption of sugar, salt, alcohol, and lower physical activity, among many other changes. These observations are nevertheless hard to reconcile with the idea that the CIM is the primary explanation for body fatness.
Just to ensure that there is no room to dispute the point I’m making here, I’ll discuss one more culture: Japan. Western Diseases contains historical data on the Japanese diet between 1950 and 1975, a time during which obesity was uncommon in Japan (p. 338). In 1975, long after postwar food shortages were over, the Japanese diet was 62 percent carbohydrate, with most of that carbohydrate coming from white rice. Furthermore, the predominant type of rice was high-amylopectin sticky rice, as it still is today (remember this word amylopectin, we’ll come back to it later). This type of rice has one of the highest glycemic loads of any food– precisely what Ludwig and Ebbeling argue is the most fattening. The Japanese ate it daily as their primary staple food, yet the prevalence of overweight and obesity were, and continue to be, lower than any other industrialized nation (19). As in many other cultures, over time the Japanese diet has become less reliant on rice and more reliant on fats, meats, sweets, and other foods, and obesity rates have increased, although they remain low compared to other countries.
Are Japanese people genetically resistant to obesity? Clearly not, because when they emigrate to the US they become much heavier (20, 21). Japanese people living in Japan are protected by their diet and lifestyle habits, despite the high glycemic load of their diet. What is different about the Japanese vs. Japanese-American diet? Many things, but here’s an obvious one: “Although total caloric intake was not greatly different between Japan and Hawaii, the percent caloric intake as fat was two times greater in Hawaii” (22). When they emigrate and their diets and lifestyles Americanize, they tend to develop overweight and obesity like people of European, African, and Native American descent, despite a reduction in dietary glycemic load.
To be clear, I’m not arguing that increased fat intake is the primary reason Japanese-Americans are heavier than Japanese, and Japanese today are heavier than in 1975– the point is to demonstrate that declining carbohydrate intake and glycemic load corresponded with an increase in body fatness. The CIM cannot be the primary explanation for obesity trends in this case, and I would argue, many other similar cases.
The genetics of common obesity argues against insulin and fat cells as the central mechanism of fat gain
Obesity has a strong genetic component, and geneticists are making great strides in cracking its code. As it turns out, the genetics of body fatness is incredibly complex, with differences at probably thousands of locations in the genome contributing to it. Each location only has a very small effect, but together they add up to a large effect. The latest and largest genome-wide analysis in 700,000 people has identified 716 places in the genome where genetic differences impact body fatness (23). By examining the functions of the implicated genes, we can gain insight into the mechanisms that determine body fatness. We know this method works: as expected, genes impacting type 2 diabetes risk tend to relate to insulin secretion, insulin sensitivity, and the pancreas, and genes impacting height tend to relate to skeletal and connective tissue growth (23, 24).
For our purposes, a key aspect of these studies is that they are unbiased (not hypothesis-driven). Researchers simply scan the entire genome looking for associations and report whatever they find, regardless of whether it conforms to our pre-existing beliefs about obesity. Whatever the mechanisms are that contribute to common obesity, they should turn up.
So what mechanisms do these studies report as being most intimately linked to obesity? Are they genes related to fat cell function? Insulin signaling? In fact, these studies consistently report that obesity-linked genes relate primarily to brain development and function: “[body mass index]-associated genes are mostly enriched among genes involved in neurogenesis and more generally involved in the development of the central nervous system” (25). This is consistent with the prevailing view in my field that the brain is in the driver’s seat, not insulin or the fat cell. That doesn’t mean the brain is the only thing that matters– probably a lot of things matter to some degree– but the brain appears to be the most important driver of common obesity. This is why my book is titled The Hungry Brain and not The Hungry Fat Cell.
I can’t overstate the importance of these genetic findings for evaluating hypotheses about obesity. They offer a rare and valuable, unbiased, 10,000-foot view of the mechanisms that drive excess body fat accumulation in the general population. We should be calibrating to these findings and asking ourselves hard questions if our hypotheses are inconsistent with them.
Circulating free fatty acids are not reduced during active fat gain in rodent models of dietary obesity
Another serious problem for the CIM is that people with obesity have circulating levels of glucose and free fatty acids (the two main circulating fuels) that are normal or elevated– not reduced as the CIM predicts (26, 27, 28, 29). Despite high levels of circulating insulin in most people with obesity, neither insulin nor anything else is preventing the release of fat from fat cells. To address this problem, Ebbeling and Ludwig conceive of a novel concept they call the “dynamic stage of obesity development”. According to this concept, during the “dynamic stage”, people are actively gaining weight because insulin is shunting fat into their fat cells, suppressing circulating fatty acids levels. But after the dynamic stage is over, weight plateaus and circulating glucose and fatty acid levels normalize or increase, which is presumably why, despite countless studies, the dynamic stage has never been observed.
Thus the CIM hinges on a phenomenon proposed by Ludwig and Ebbeling that has never been observed, and that honestly I find a bit too convenient. But has it been disproven? Let’s see if we can find any data. What we want are time course studies where researchers measure circulating free fatty acid levels at regular intervals in humans or animals that are actively gaining fat. I found two studies in rats, both of which report that circulating free fatty acids were normal or elevated at all points during active fat gain resulting from a fattening diet (30, 31). In this model of obesity at least, there is no “dynamic stage of obesity development” that supports the predictions of the CIM. Importantly, obesity was caused by a fattening diet in these studies. This is more pertinent to common human obesity than studies that involve brain lesions, leptin deficiency, or excess insulin injections, which tend to be cited by Ludwig and Ebbeling.
Reducing the flow of fatty acids out of fat cells doesn’t reduce energy expenditure, increase food intake, or increase body weight in humans
The cornerstone of the CIM is the idea that insulin suppresses fat release from fat cells, reducing energy levels in the blood, which reduces metabolic rate (energy expenditure), increases hunger, and causes fat gain. What if there were a way to experimentally restrict the flow of fatty acids out of fat cells, independently of insulin? If the CIM is correct, we should see a decrease in energy expenditure, an increase in food intake, and fat gain.
Fortunately for us, there is a way to do this: a drug called acipimox. Acipimox inhibits lipolysis, or the release of fatty acids from fat cells, mimicking the effect of insulin (32). As a consequence, free fatty acid levels in circulation decline. Acipimox has been used in humans many times, but of particular interest to us is a six-month randomized controlled trial that reported the impact of acipimox vs. placebo on energy expenditure, food intake, and body composition (33). The (preregistered) primary goal of this study was to examine the effects of acipimox on mitochondrial function, but it reported a number of other outcomes (34).
Researchers randomly assigned 39 men and women with obesity to acipimox or placebo for six months. 31 continued for the whole 6-month period (16 in acipimox and 15 in placebo) and compliance was excellent. Acipimox robustly and consistently inhibited lipolysis and at the six-month time point fasting free fatty acids in circulation were reduced by 38 percent.
How did energy expenditure change? “No significant effects of acipimox compared to placebo on [resting energy expenditure] or [respiratory quotient] were observed during either the fasting state or the hyperinsulinemic clamp.” And: “Changes in physical activity over 6 months were not different between groups”
How about food intake? According to 4-day food records: “Caloric and relative macronutrient intake did not change significantly between groups”.
Body composition? To measure this, they used the accurate DEXA technique: “There were no significant effects of acipimox vs placebo on measures of body composition, including [body mass index], [visceral adipose tissue], or lean body mass”.
Acipimox simulated the effect of insulin on fat cells that is supposed to make us hungry and fat according to the CIM, but it had no effect on food intake or body composition. Furthermore, this study suggests that the brain doesn’t respond to low free fatty acid levels by increasing food intake, which is another unproven assumption that underlies the CIM. These findings are very hard to reconcile with the CIM.
Animal studies cited by Ebbeling and Ludwig are not as supportive as suggested
Ludwig and Ebbeling cite rodent studies their team and others conducted in which they placed rats and mice on low-glycemic/insulinemic vs. high-glycemic/insulinemic diets. Since they rely heavily on these rodent studies in their arguments, and ostensibly believe rodents are useful animal models for human obesity, let’s take a closer look at them.
Let’s start with a series of studies, conducted by Gerard Slama and colleagues between 1996 and 1998, cited by Ludwig and Ebbeling. These compared diets based on mung bean noodles (low glycemic) or ground-up toast (high glycemic) in rats. You read that correctly: mung bean noodles vs. powdered toast. According to the authors, mung bean noodle starch is 8 percent indigestible fiber (resistant starch) while ground-up toast starch is only 2 percent indigestible fiber (35). So right off the bat, these diets were not isolating the glycemic index as a variable– they also differed substantially in fiber content, somewhat in calorie density, and probably in many other ways. Furthermore, resistant starch is a type of fiber that is actively under study for its potential weight and health benefits.
In the first study, published in 1996, the researchers put diabetic and nondiabetic rats on the two diets for five weeks (36). I’ll let them share the results in their own words: “Body weight was significantly lower in rats (normal and diabetic) fed on the mung-bean starch diet until the fourth week. However, body weight was comparable for both diets at the end of the 5-week period.” I don’t see how this supports the CIM.
Onward to the second study, published in 1998 (37). Again, they put diabetic and nondiabetic rats on the noodles vs. toast diets, this time for three weeks. The abstract says what we want to know: “After 3 wk, food intake, epididymal fat pad weights, and plasma glucose, insulin and triglyceride concentrations did not differ between diet groups.” Again, I don’t see how this supports the CIM.
The third study was published later in 1998 (38). Same design and the previous study, same findings: “After 3 wk, neither body weights nor relative epididymal fat pad weights differed.” None of the three studies cited by Ludwig and Ebbeling found that noodles vs. toast meaningfully impacts weight or body fatness in rats, despite other differences in diet composition that should have favored the low-glycemic noodle diet.
Let’s move on to a study conducted by Ludwig’s group and published in 2004 (39). It’s much better controlled than the noodles-vs.-toast studies, as the only difference between the two diets was the type of starch: fast-digesting amylopectin vs. slow-digesting amylose. In the first experiment, the researchers began by removing 60 percent of the rats’ pancreas to impair their insulin secretion. I don’t buy the rationale for doing this (wouldn’t reducing insulin secretion make carbohydrate less fattening according to the CIM?); they say it’s to make the rats more similar to people with prediabetes. Rats were fed the diets for 18 weeks. The amylopectin group began gaining weight halfway through and the researchers had to restrict their food intake to keep their weight similar to the amylose group. Despite no differences in weight and a lower calorie intake, the amylopectin group ended up fatter than the amylose group at the end, although neither group was especially fat (18 vs. 10 percent body fat).
I’m going to skip over the second experiment because it isn’t very relevant here. In the third experiment, they fed normal obesity-prone mice (C57BL/6J strain) the two diets for 9 weeks without restricting their intake. Body weight didn’t differ at the end, but body fatness was meaningfully higher in the amylopectin group (25 vs. 14 percent body fat). A longer-term study by Ludwig’s group also showed that an amylopectin-based diet leads to a meaningfully higher level of body fatness than an amylose-based diet in mice (40).
Together, these experiments do suggest that amylopectin is more fattening than amylose in rodents, although amylopectin-fed rodents look like runway models next to those fed refined high-fat diets (as described above). However, it’s strange to me that they only reported results from rats that had 60 percent of their pancreas removed. No matter, because I was able to find another study that put normal rats on amylose or amylopectin diets for 16 weeks (41).
This study is really interesting because it not only included amylose and amylopectin-based diets, it also included a third glucose-based diet. Glucose is digested and absorbed even more quickly than amylopectin, and stimulates insulin to a correspondingly greater degree. So if the glycemic index is what really matters, we should see the most weight gain in the glucose group, followed by the amylopectin group, followed by the amylose group. Here’s what happened: “There was no significant difference in body weight between the [amylose] or [amylopectin] groups at any of the testing points. The [glucose]-fed animals gained weight at a slightly lower rate than the [amylose] or [amylopectin]-fed animals. The [glucose]-fed animals had a significantly lower body weight than [amylose]- or [amylopectin]-fed animals at both the 8- and 16-wk testing points.” This is despite elevated insulin secretion in the glucose group.
The highest-glycemic diet caused the least weight gain, and there was no difference between the amylopectin and amylose groups eating ad libitum, in contrast to Ludwig’s study where they had to restrain the amylopectin-fed rats’ food intake to rein in body weight. It’s worth repeating that 16 weeks of a refined high-fat diet causes severe, unmistakable obesity in rodents (42, 43).
This leads me to question whether glycemic/insulinemic index is the relevant difference between amylose and amylopectin-based diets. If that were the case, wouldn’t a glucose-based diet be the most fattening of all? Another thing that leads me to question the relevance of these findings is the human evidence. Low-glycemic/insulinemic diets are not very effective for fat loss in humans, including in Ludwig and Ebbeling’s randomized controlled trial where their low-glycemic diet caused weight loss nearly identical to a low-fat diet (44). Then there are the Japanese data I discussed above, where the consumption of amylopectin-rich sticky white rice as the primary staple food did not lead to obesity (Western Diseases: Their Emergence and Prevention).
Is the CIM well-enough supported to justify diet advice?
Ludwig and Ebbeling state that “high-quality research will be needed to resolve the debate”, yet their paper contains a panel titled “Dietary Recommendations Based on the Carbohydrate-Insulin Model”. As I recall, one of the favorite pastimes of CIM advocates is criticizing the USDA for prematurely dispensing low-fat diet advice in the 1980s. If the science isn’t settled yet, perhaps it’s not the right time for bestselling books that confidently promote the CIM and a diet based on it to the public?
Conclusions
The question we must answer is not “can we find evidence that supports the CIM”, but rather “does the CIM provide the best fit for the totality of the evidence”. Although it is certainly possible to collect observations that seem to support the CIM, the CIM does not provide a good fit for the totality of the evidence. It is hard to reconcile with basic observations, has failed several key hypothesis tests, and currently does not integrate existing knowledge of the neuroendocrine regulation of body fatness.
Certain forms of carbohydrate probably do contribute to obesity, among other factors, but I don’t think the CIM provides a compelling explanation for common obesity.

 I recently heard the sad news that Staffan Lindeberg, MD, PhD, lead researcher of the Kitava Study, has died.
I recently heard the sad news that Staffan Lindeberg, MD, PhD, lead researcher of the Kitava Study, has died.